
1. はじめに
「あなたのルーツはどこですか?」と聞かれて、私たちは自分の家系や出身地を思い浮かべます。しかし、医療の現場で本当に必要なのは、遺伝子そのものに刻まれた「生物地理学的な祖先」の情報です。
がんの治療は、いまや「全員に同じ薬」から「その人の腫瘍の遺伝的特徴に合わせた薬」へと舵を切りました。包括的ゲノムプロファイリング(Comprehensive Genomic Profiling, CGP)は、ひとつの検査で数百もの遺伝子変異を一度に読み取り、患者ごとの最適な治療を選ぶための地図を描く技術です。
ところが、ここに大きな落とし穴がありました。これまでの臨床研究のほとんどは、患者の人種を「自己申告」に頼ってきたのです。「白人」「アジア人」「ヒスパニック」といった大雑把な区分は、実際の遺伝子の多様性を表しきれません。たとえば「ヒスパニック」と一口に言っても、その遺伝的背景はヨーロッパ系・先住民系・アフリカ系が混ざり合い、人によってまったく異なります。
この溝を埋めるために登場したのが、腫瘍DNAから遺伝的祖先(Genetically Inferred Ancestry, GIA)を推定する技術です。それも、健康な細胞のDNAを別に採らずに、がんの検査で得たCGPデータだけから推定する。しかも、複数のAI分類器を組み合わせ、答えを出す。そんな仕組みが特許と論文の両面で立ち上がりつつあります。
本記事では、この腫瘍DNAからの祖先推定という、精密医療と健康格差解消の交差点に立つ次世代技術を、特許3件と最新文献から読み解きます。
2. 腫瘍DNAから「祖先」を読み取るとは
2-1. なぜ自己申告では足りないのか
医療研究で長年使われてきた「自己申告人種・民族(Self-Identified Race and Ethnicity, SIRE)」は、調査票に並ぶ数個のカテゴリから選ぶ仕組みです。便利な反面、遺伝的な実態とのズレが大きいことが知られています。
たとえば、ある患者が「白人」と回答しても、その遺伝子には北アフリカ系や中東系の祖先成分が含まれることがあります。薬の代謝に関わる遺伝子変異の頻度は集団ごとに大きく異なるため、この「ズレ」は治療選択を狂わせかねません。Labcorp Oncologyらの研究グループは、SIREには「普遍的な基準がなく、広すぎる人種・民族カテゴリーに依存している」と指摘し、遺伝的祖先こそが患者の実像を映す客観的な指標だと述べています。
2-2. 「腫瘍だけ」で祖先を推定する難しさ
ここで難題が立ちはだかります。CGP検査は基本的に腫瘍のDNAを読み取るためのもので、健康な細胞のDNA(マッチドノーマル)を一緒に採らないケースが多いのです。
しかし腫瘍DNAには、がん細胞特有の体細胞変異(後天的な変異)が大量に含まれています。これが祖先推定のノイズになります。本来、祖先推定で見たいのは生まれつき持っている変異(生殖細胞系列のSNP)であり、がん化の過程で生じた変異は邪魔者なのです。
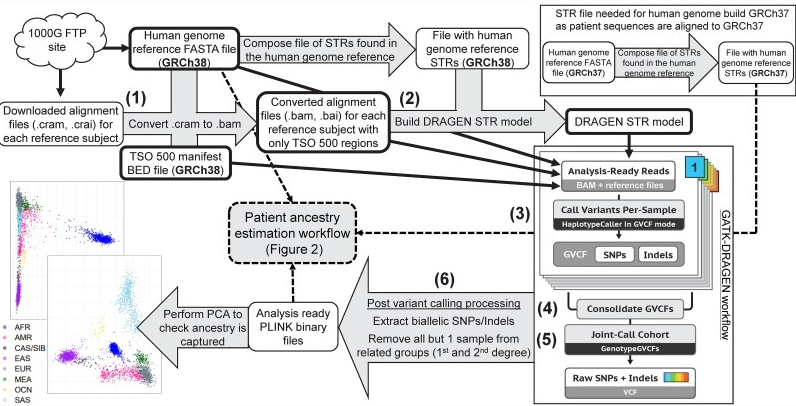
この問題に対し、AACR Cancer Researchに掲載されたBelleau らの研究は、「がん由来の分子データだけから祖先を高精度に推定できる」計算手法を提示しました。腫瘍シーケンスから生まれつきの変異だけを抽出して再構成する流れは、現在のGIA技術の前提となっています。
2-3. リファレンスパネルという「世界地図」
祖先を推定するには、比較対象となる「世界の遺伝子地図」が必要です。標準として使われているのが1000 Genomesプロジェクトで、世界26集団の数千人分のゲノムデータが公開されています。
近年は、これにヒトゲノム多様性プロジェクト(HGDP)やSimons Genome Diversity Project(SGDP)が加わり、中央アジア・シベリア、中東、オセアニアといった、これまでカバーが薄かった集団も補強されました。患者の腫瘍DNAから抽出したSNPを、この世界地図の上にプロットすることで、「あなたの遺伝子はこの集団に近い」という座標が描けるのです。
3. 特許から見る技術革新
3-1. 特許①:祖先情報マーカー(AIMs)という「設計図」
腫瘍DNAから祖先を読むための土台となるのが、祖先情報マーカー(Ancestry Informative Markers, AIMs)という考え方です。集団ごとに頻度が大きく異なるSNPを選び抜き、その組み合わせで集団構造を推定するアプローチで、Tony FrudakisとMark Shriverらが2003年に出願した特許US20040229231A1「Compositions and methods for inferring ancestry」は、この分野の基礎を築きました。
この特許は、「少なくとも約10個以上のAIMsを含むSNPパネルでハイブリダイゼーション検出を行い、所定の信頼水準で集団構造を同定する」という方法を請求しています。注目すべきは、検出するAIMの数が増えるほど推定の信頼度が上がるという定量的な枠組みを示した点です。現代のCGPパネルでは数万から数十万のSNPが副次的に読み取れるため、この特許の発想が大幅に拡張された形で生きています。
3-2. 特許②:祖先特化型リスクスコアという「処方の地図」
祖先が分かったところで、それをどう使うのか。Asia Genomics Pte LtdのUS10468141B1「Ancestry-specific genetic risk scores」は、推定された祖先をリスク予測の精度向上に直結させる仕組みを提示しました。
この特許の核は、集団ごとの連鎖不平衡(Linkage Disequilibrium, LD)パターンを使うことにあります。ある疾患に関連する遺伝子変異が患者で直接観測できなくても、同じ集団内でLD関係にある別の変異(プロキシ変異)から補完できる、という発想です。
ここでも祖先推定はPCA(主成分分析)または最尤推定(MLE)で行うと明記されています。つまり、祖先を「分類」しただけでは終わらせず、その人専用のリスクスコアを動的に組み立てるところまで一気通貫で設計されている点が技術的に新しいのです。たとえばアフリカ系で頻度が高いLDブロックと、東アジア系で頻度が高いLDブロックを取り違えると、リスクスコアは大きく狂います。それを自動的に補正する仕組み、と言ってよいでしょう。
3-3. 特許③:cfDNAと個別化パネルが拓くリキッドバイオプシー
3つ目は、Myriad Womens Health IncのUS12344901B2「Automated methods of detecting cell free DNA」です(2025年7月成立)。
この特許は、患者の腫瘍組織からまず個別化シグネチャーパネル(その患者の腫瘍だけで陽性となる体細胞変異の集合)を作り、その後は血液10〜20mLという少量のサンプルから循環腫瘍DNA(ctDNA)を超高感度で検出する、という二段構えの仕組みを請求しています。
注目したいのは、二本鎖シーケンス(デュプレックスシーケンシング)と分子バーコードを組み合わせてランダムな配列誤りを除去する点と、この技術が出生前検査や移植モニタリングにも応用可能だと明記されている点です。つまり、本記事の起点となるCGP × GIA技術が「腫瘍・移植・出生前」という3つの臨床応用に橋渡しできる、その血管に当たる基盤特許なのです。
4. 応用分野・実用化
4-1. がん精密医療:薬の選び方が変わる
最も直接的な応用は、がん治療薬の選択です。EGFR変異や BRCA1/2変異の頻度は集団によって大きく異なり、効果の出やすい薬や副作用のリスクも変わります。Foundation MedicineがNEJMで報告した研究では、米国50州から620,500サンプルを9.5年間にわたり解析し、アフリカ系祖先の患者比率が年0.5ポイントずつ増加して2022年9月に12.4%に達したと示しました。GIAは、これまで研究データから抜け落ちがちだった集団に、適切な治療機会を届ける道具になりつつあります。
4-2. 移植医療:ドナーとレシピエントの「距離」を測る
臓器移植では、HLA型の一致だけでなく、両者の遺伝的距離が拒絶反応に影響します。CGPで得たSNP情報からGIAを推定し、ドナー・レシピエントの祖先構成を客観的に比較すれば、リスク評価が精緻になります。さらに、特許3で触れたcfDNA技術と組み合わせれば、移植後の血液から「ドナー由来DNAの増減」をモニタリングし、拒絶の早期発見にもつながります。
4-3. 出生前診断:母体血からの読み取り
非侵襲的出生前検査(NIPT)でも同じ枠組みが使えます。母体血中には胎児由来のcfDNAが循環しており、ここから胎児の染色体異常を読み取る検査がすでに普及しています。GIA技術が組み込まれれば、胎児の遺伝的祖先を踏まえた集団特有の遺伝病リスクを、より正確に評価できるようになります。
5. 課題と展望
5-1. 「人種カテゴリー」の限界をどう超えるか
現在のGIA技術が最終的に出力するのも、結局は「アフリカ系」「ヨーロッパ系」「東アジア系」といった大陸レベルの分類が中心です。Labcorpの論文では8集団(アフリカ、混合アメリカ、中央アジア/シベリア、ヨーロッパ、東アジア、中東、オセアニア、南アジア)に拡張されましたが、それでも個人の混合度合いまで完全に表現するには足りないという課題があります。
5-2. 研究の最前線:3つの分類器による「合議制」
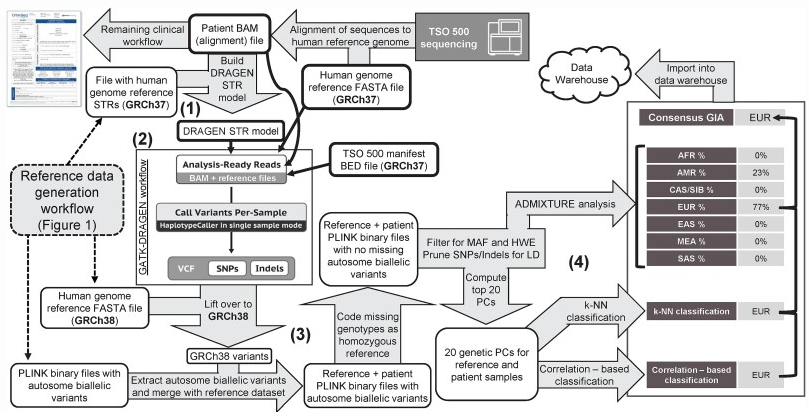
最新の流れは、コンセンサス型分類です。Labcorpの実装では、上位20の遺伝的主成分(PC)を入力として、k近傍法(k=8)、相関ベースのカスタム分類器、そしてADMIXTUREによる混合解析という3つの異なる方法で祖先を判定し、その合議で最終結論を出します。
この設計の強みは、ひとつの手法が苦手とする集団(たとえば混合集団)でも、他の手法が補ってくれる点にあります。実際、4274人の参照サンプルと491人の患者で検証したところ、SIREとの一致率は95%、患者全体での祖先割り当て成功率は99.4%(501/504)に達しました。
AACR Project GENIEも同様の方針で、NGSパネルのオフターゲットリード(標的外領域のシーケンス)から祖先をインピュテーションする基盤を構築中だと公表しています。検査の主目的とは別に、副産物として祖先情報まで取れる時代が来ているのです。
5-3. 未来の展望:データ駆動型の健康格差解消
2025年にNature Geneticsで報告されたメタ解析は、遺伝的祖先によってがんの体細胞変異プロファイルに明確な差があることを示しました。これは「人種で薬を選ぶ」という古い発想を超え、集団遺伝学のレイヤーから治療戦略を組み直すことの必要性を裏づけています。
将来的には、CGP検査を受けるたびに、患者ごとに「祖先プロファイル」「変異プロファイル」「祖先特化リスクスコア」がワンセットで出力されるようになるでしょう。それは単なる医療データの拡張ではなく、これまで研究から取り残されてきた集団を含めた、真の意味での精密医療への道筋です。
あわせて読みたい
「ゲノムから個人を読み解く」という同じ流れで、シーケンスデータが薄くても遺伝情報を補完する仕組みに興味のある方には、こちらの記事が参考になります。

「リスクを数値で語る精密医療」の別の切り口として、タンパク質情報から疾患リスクを予測する技術を扱った記事もあわせてどうぞ。

6. 結論
CGPは、もはや「がんの変異を読む」だけの技術ではありません。同じシーケンスデータから、患者の遺伝的ルーツまでを読み取り、それを治療選択に反映させる。そんな包括的なプラットフォームへと進化しています。
その中心にあるのが、腫瘍DNAから祖先を推定するコンセンサス型AIの仕組みです。AIMsという基礎特許、祖先特化型リスクスコアという応用特許、そしてcfDNA検出の周辺特許。これらが互いに支え合いながら、「人種ではなく祖先で語る精密医療」という新しい標準を形づくっています。
一見すると地味な統計学とゲノム科学の融合領域に、次世代医療の最も重要なフロンティアの一つが眠っているのです。
関連アイテム
ゲノムが語る人類全史(アダム・ラザフォード 著/垂水雄二・篠田謙一 訳/文藝春秋)
DNA解析が拓く「祖先と人類史」の世界が、ダイナミックに描かれた本です。本記事のGIA技術が依拠する集団遺伝学の発想を、文学的な読み味で体感できる一冊です。
参考文献
テーマに近い関連する特許文献
- Frudakis, T. & Shriver, M. Compositions and methods for inferring ancestry. US20040229231A1 (DNAPrint Genomics Inc / DNA Diagnostics Center Inc, 2004). https://patents.google.com/patent/US20040229231A1/en
- Valenzuela, R. K. et al. Ancestry-specific genetic risk scores. US10468141B1 (Asia Genomics Pte Ltd, 2019). https://patents.google.com/patent/US10468141B1/en
- Maguire, J. R. et al. Automated methods of detecting cell free DNA. US12344901B2 (Myriad Womens Health Inc, 2025). https://patents.google.com/patent/US12344901B2/en
記事を作成するにあたり参考にした文献
- Wallen, Z. D. et al. A consensus-based classification workflow to determine genetically inferred ancestry from comprehensive genomic profiling of patients with solid tumors. Briefings in Bioinformatics, 25(6), bbae557 (2024). https://pmc.ncbi.nlm.nih.gov/articles/PMC11521331/
- Belleau, P. et al. Genetic Ancestry Inference from Cancer-Derived Molecular Data across Genomic and Transcriptomic Platforms. Cancer Research, 83(1), 49–58 (2023). https://aacrjournals.org/cancerres/article/83/1/49/711844/Genetic-Ancestry-Inference-from-Cancer-Derived
- Sweeney, S. M. et al. Addressing racial and ethnic disparities in AACR project GENIE. npj Precision Oncology, 7, 81 (2023). https://www.nature.com/articles/s41698-023-00425-5
- Mata, D. A. et al. Disparities According to Genetic Ancestry in the Use of Precision Oncology Assays. New England Journal of Medicine / Foundation Medicine Research Spotlight (2023). https://www.foundationmedicine.com/research/research-spotlight-disparities-according-genetic-ancestry-use-precision-oncology-assays
- The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature, 526, 68–74 (2015). https://www.nature.com/articles/nature15393
- International Genome Sample Resource (IGSR). 1000 Genomes — A Deep Catalog of Human Genetic Variation. https://www.internationalgenome.org/
- Arora, K. et al. Meta-analysis reveals differences in somatic alterations by genetic similarity of populations in cancer. Nature Genetics (2025). https://www.nature.com/articles/s41588-025-02371-3
コメント